Journal of Molecular Evolution (1998) 46, 84-101
Evolution of substrate specificities in the P-type ATPase superfamily
Kristian B. Axelsen1,2,*,
Michael G. Palmgren1
1 Department of Plant Physiology,
Institute
of Molecular Biology, Copenhagen University, Øster Farimagsgade 2A, DK-1353 Copenhagen K, Denmark
2Department of Plant Biology, Royal Veterinary and Agricultural University, Thorvaldsensvej 40, DK-1871 Frederiksberg C, Denmark
*Correspondence should be addressed to K.B.A.
e-mail kax[at]life.ku.dk;
ABSTRACT
P-type ATPases make up a large superfamily of ATP-driven pumps involved in the trans-membrane transport of charged substrates. We have performed an analysis of conserved core sequences in 159 P-type ATPases. The various ATPases group together in five major branches according to substrate specificity, and not according to the evolutionary relationship of the parental species, indicating that invention of new substrate specificities are accompanied by abrupt changes in the rate of sequence evolution. A hitherto unrecognized family of P-type ATPases has been identified that is expected to be represented in all the major phylae of eukarya.
Key words: Na+ - K+ - Cu2+ - Ca2+ - H+ - Mg2+ - Cd2+ - Phospholipid translocases
INTRODUCTION
P-type ATPases comprise a ubiquitous family of proteins involved in the active pumping of charged substrates across biological membranes (Møller et al. 1996). Their distinguishing feature is the formation of a phosphorylated intermediate during the reaction cycle (hence P-type). P-type ATPases of various substrate specificities have several functions. In animals they provide, for example, the basis for action potentials in nervous tissues, secretion and reabsorption of solutes in the kidneys, acidification of the stomach, nutrient absorption in the intestines, relaxation of muscles, and Ca2+ dependent signal transduction.
Several reports have speculated about the relationships among the various P-type ATPases (Serrano 1988; Fagan and Saier 1994, Møller et al. 1996) but the number of proteins included in these analyses have been relatively low. Since the last detailed investigation (Fagan and Saier 1994), at least one new class of P-type ATPases has been discovered (Auland et al. 1994; Tang et al. 1996), systematic sequencing efforts have provided the complete sequences of several genomes (Fleischmann et al. 1995; Fraser et al. 1995; Bult et al. 1996; Goffeau et al. 1997; Himmelreich et al. 1996; O'Brien 1997) and a substantial amount of sequence information has been obtained from other organisms. This has made it possible to analyse the P-type ATPase content of living organisms from all three domains of life and to establish the presence of five major groups of P-type ATPases.
METHODS
Database Searches
The EMBL database (release 51), SWISS-PROT (release 34) and expressed sequence tag (EST) databases (EMEST and GBEST, until December 15 1996) were screened using the FASTA, TFASTA and BLAST programs of the Wisconsin package, version 8 (Program Manual for the Wisconsin Package, Version 8 September 1994 Genetics Computer Group, 575 Science Drive, Madison, Wisconsin, USA 53711 (1994). The genomes of Mycoplasma genitalium, Haemophilus influenzae, and Methanococcus jannaschii were accessed via the world wide web at The Institue for Genome Research, the genome of Synechocystis PCC6803 at the Kazusa DNA Research Institute (KDRI), and the genome of Escherichia coli was accessed at the E. coli Genome Project at University of Wisconsin-Madison and at the E. coli Databank.
Identification of P-type ATPase Sequences
The initial screen of the EMBL and SWISS-PROT databases was performed with the PROSITE consensus sequence for P-type ATPases DKTG[T,S][L,I,V,M][T,I]. All sequences but 13 obtained matched the sequence DKTGTLT, 12 sequences matched DKTGTIT (six mammalian and two bacterial heavy metal transporting-ATPases, three bacterial KdpB ATPases involved in the transport of K+, and the CTA3 ATPase of Schizosaccharomyces pombe believed to transport Ca2+ and one matched DKTGTII (a human Cu2+-ATPase; Bull et al. 1993). All matches contained in addition most or all of ten conserved sequence motifs previously identified in a number of P-type ATPases (Serrano 1988; Møller et al. 1996). Several of these conserved regions were used to re-screen the databases to find possible P-type ATPases which do not match the DKTGT[L,I][T,I] sequence. Using this method, no further sequences encoding putative P-type ATPases were found. The screening of the databases revealed a total of 211 independent full length open reading frames encoding P-type ATPases.
Construction of Phylogenetic Trees
Alignments of the sequences were performed with the clustalw program (Thompson et al. 1994). Phylogenetic analysis were performed with the Protdist and Fitch (Fitch-Margoliash and least squares method) programs of the Phylip package (Felsenstein 1989). To make sure that the phylogenetic relationship found was not skewed because only partial sequences were used in the analysis, the full length proteins of the members of the different families were aligned and the alignments were inspected for similarities outside the core regions. The closer relationship between members of the families resulting from the phylogenetic analysis could be confirmed by these alignments.
Analysis of the Expressed Sequence Tag (EST) Databases
The GBEST and EMEST databases as well as the TIGR non-redundant EST database were screened with all the known cDNA sequences and the coding region of genomic sequences encoding P-type ATPases in the organisms was analysed. In this way ESTs identical to already cloned P-type ATPases in that organism were identified. In addition, 25 full-length protein sequences from all branches of the evolutionary tree in Fig. 6 were searched against the EST databases. EST sequences that showed at least 20 % sequence identity within a stretch of 30 amino acid residues to any one sequence were translated and searched against all databases. EST sequences showing significant homology to P-type ATPases only were subsequently extracted. These EST sequences were compared to the sequences of all cloned P-type ATPases. In this way the P-type ATPase to which each of the EST sequences had the closest resemblance was identified.
RESULTS
Conserved Core Sequences in P-type ATPases
A total of 211 P-type ATPase sequences were identified in the databases. Many sequences showed less than 15% identity to each other and varied considerably in length; from 642 amino acid residues in a heavy metal (HM) ATPase from Synechocystis PCC6803 (seq. 27 in Table 1; Fig. 1) to 1956 residues in a gene from Plasmodium falciparum of unknown function (seq. 4 in Table 1; Fig. 1). Due to large differences in primary structure and low overall similarity, a complete multiple alignment of all full length P-type ATPases proved to be an impossible task. However, we identified in the partial multiple alignment eight regions (A - H; Figs. 2 and 3), comprising a total of 265 amino acids that always aligned and in which only minimal deletions and insertions were found. The KGAP motif found in a variety of P-type ATPases (Serrano 1988) can not be unambiguously detected in HM-ATPases (Møller et al. 1996) and was not considered a core region.
The eight conserved regions, defining the core of the P-type ATPase superfamily, were extracted from the various P-type ATPases and arranged in a linear sequence (Fig. 1). The number of sequences was subsequently reduced to avoid overrepresentation of closely related sequences. In the resulting dataset, containing 159 core sequences, no P-type ATPase represented was more than 90% identical (when comparing full-length proteins) to any other member of the set. The excluded P-type ATPases are listed in Table 2.
A Phylogenetic Tree of P-type ATPases
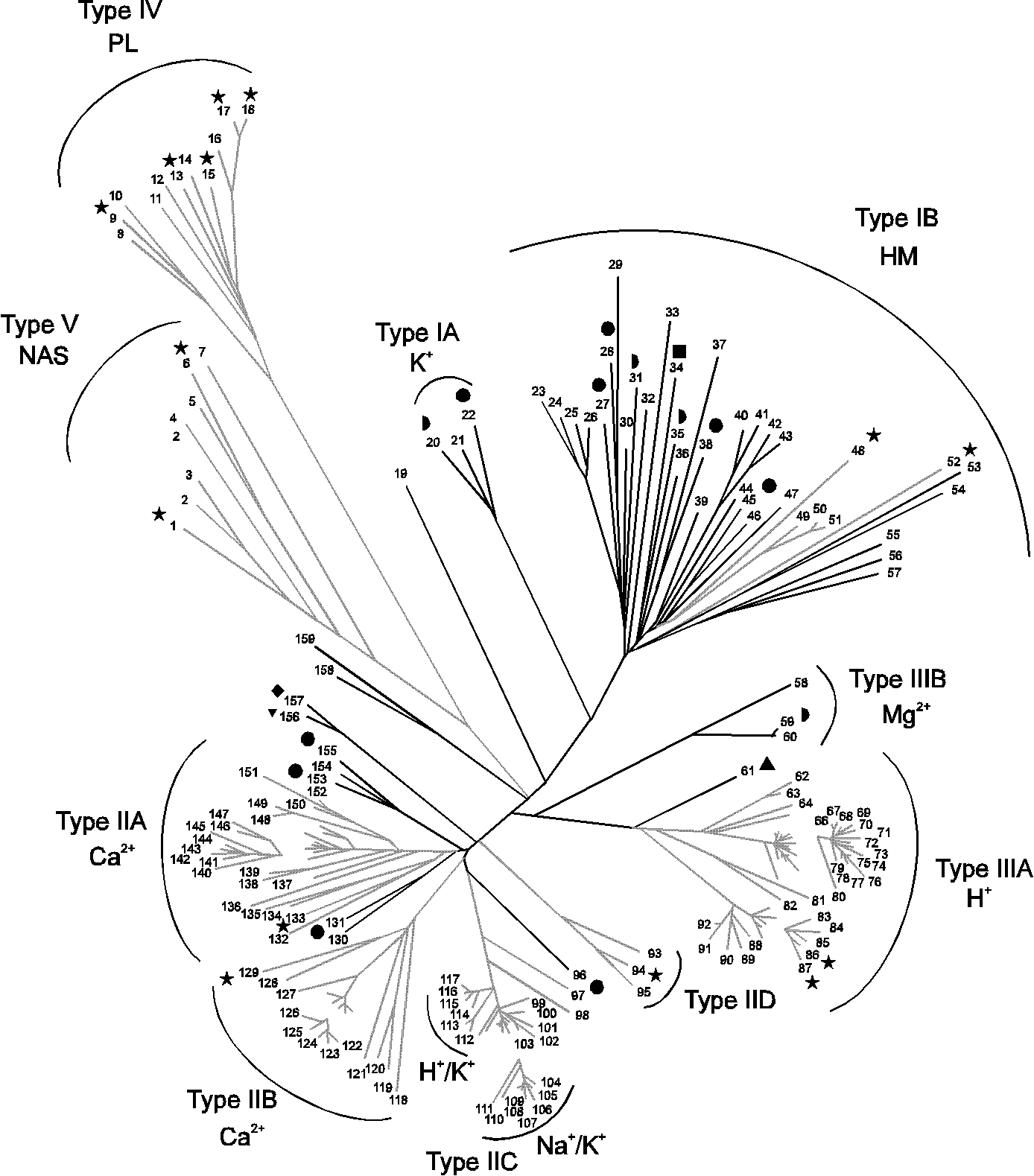
A total of 14 phylogenetic trees were produced using the same dataset of sequences, but presenting them in different orders. Among these, 12 were identical with respect to the position of the major branches of the tree. A simplified version of the different trees obtained is shown in Fig. 4. All trees reveal five main branches, designated Type I - V ATPases, with a number of minor families. Amino acids within core segments conserved in the various branches of P-type ATPases are indicated in Fig. 5. The main difference between the two types of trees presented in Fig. 4 is the position of Type V ATPases. Although Type IV and Type V ATPases group together in most trees, this is not always the case, and therefore these groups were not considered to belong to the same type. In contrast, Type IA and IB ATPases as well as Type IIIA and IIIB ATPases always grouped together and were therefore considered to be of monophyletic origin and belonging to the same type. The phylogenetic tree that appeared most times (12 times out of 14) is shown in Fig. 6 and is discussed below.
The first main branch (Type IB ATPases) encompasses HM-ATPases and constitutes a large, rather distantly related group of proteins. It includes at least two families, one of which contains all known Cu2+-ATPases and the other includes four putative Cd2+-ATPases (seq. 23-26; Fig. 6). The KdpB proteins of Escherichia coli, Synechocystis PCC6803, and Mycobacterium tuberculosis (seq. 20-22; Fig. 6), all of which are involved in K+ transport, form a family of their own (Type IA ATPases) that is closely related to HM-ATPases.
The second branch (Type II ATPases) is split into several families exhibiting various levels of conservation. Most ATPases in this branch are believed to transport Ca2+ and are divided into two relatively distant families (Type IIA and Type IIB ATPases). Type IIA ATPases also include five putative Ca2+-ATPases from three bacterial species that form two separate groups in this family (seq. 130-131 and 153-155; Fig. 6). Two families, Na+/K+ and H+/K+-ATPases, are very closely related and are designated Type IIC ATPases. In addition, a small family of fungal ATPases (Type IID ATPases) and the solitary P-type ATPases in Mycoplasma genitalium and Mycoplasma pneumoniae of unknown function (seq. 156-157; Fraser et al. 1995; Himmelreich et al. 1996) form separate groups in the branch of Type II ATPases.
The third branch (Type IIIA ATPases) covers plasma membrane H+-ATPases. A small group of Mg2+-ATPases from two bacterial species (Type IIIB ATPases) group together with the H+-ATPases.
The fourth branch (Type IV ATPases) constitutes a recently discovered family of enzymes found only in eukarya, some of which (e.g. seq. 14; Table 1) have been shown to be involved in the transport of aminophospholipids (Auland et al. 1994; Tang et al. 1996). A group of P-type ATPases (Type V ATPases) having no assigned specificity constitutes the fifth branch of the phylogenetic tree. This distinct group of eukaryotic ATPases has three members from Caenorhabditis elegans, two members from Saccharomyces cerevisiae and one member each from P. falciparum and Tetrahymena thermophila. A characteristic sequence feature of the Type V ATPases is the PPxxP motif in region D (Fig. 5). These pumps have not been identified as a group before.
Number and Nature of P-type ATPases in Fully Sequenced Bacteria and Archaea
Synechocystis PCC6803, Escherichia coli and Mycobacterium tuberculosis are phylogenetically rather distant bacterial species belonging to cyanobacteria, proteobacteria and Gram-positive bacteria, respectively (Olsen et al. 1994). Still, they have a comparable profile and number of P-type ATPases: nine and four genes are found in the fully sequenced bacteria Synechocystis PCC6803 and E. coli, respectively, and ten P-type ATPases are cloned from the partially sequenced bacterium M. tuberculosis (Table 3). This suggests that a common trait for bacteria is having several P-type ATPases. The P-type ATPases of these organisms fall into three main classes: Type IA ATPases (one each in the three organisms; Fig. 6), Type IB ATPases (five in M. tuberculosis, four in Synechocystis, and two in E. coli), and Type IIA ATPases (one in M. tuberculosis and three in Synechocystis). A number of bacterial P-type ATPases have no or very few close relatives among other ATPases and thus form their own branches. This is true for three P-type ATPases from M. tuberculosis (function unknown; seq. 19, 158, and 159; Fig. 6), the Type IIIB ATPases of E. coli and Salmonella typhimurium (seq. 58-60; Fig. 6), and one of the P-type ATPases from Synechocystis (seq. 96; Fig. 6), which is most closely related to Type IIC ATPases.
Haemophilus influenzae, although very closely related to E. coli (both are in the 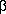 subdivision of proteobacteria; Olsen et al. 1994) only has a single P-type ATPase (Table 3). The same is true for M. genitalium and M. pneumoniae (Table 3), two very closely related Gram-positive bacteria. The solitary P-type ATPases in H. influenzae and M. genitalium (or M. pneumoniae) exhibit very low similarity to each other and group in distinct families of the phylogenetic tree of ATPases (Type IB ATPases and Type II ATPases, respectively; Table 3; Fig. 6). H. influenzae, M. genitalium and M. pneumoniae live in humans in constant environmental conditions. This may explain why some bacteria in special niches can do without certain P-type ATPases and might have eliminated superfluous ATPases with time.
subdivision of proteobacteria; Olsen et al. 1994) only has a single P-type ATPase (Table 3). The same is true for M. genitalium and M. pneumoniae (Table 3), two very closely related Gram-positive bacteria. The solitary P-type ATPases in H. influenzae and M. genitalium (or M. pneumoniae) exhibit very low similarity to each other and group in distinct families of the phylogenetic tree of ATPases (Type IB ATPases and Type II ATPases, respectively; Table 3; Fig. 6). H. influenzae, M. genitalium and M. pneumoniae live in humans in constant environmental conditions. This may explain why some bacteria in special niches can do without certain P-type ATPases and might have eliminated superfluous ATPases with time.
The archaeon Methanococcus jannaschii only possesses a single P-type ATPase showing high similarity to Type IIIA ATPases. Whether the simple genome of M. jannaschii with respect to P-type ATPases is an exception among archaea cannot be determined before more archaean genomes have been sequenced.
P-type ATPases in the Yeast Saccharomyces cerevisiae
As many as 16 P-type ATPase genes are found in the genome of the unicellular yeast S. cerevisiae (Table 1, 2, and 3). These belong to all the five main branches of the phylogenetic tree (Fig. 6). Single yeast sequences belong to Type IIA and IIB Ca2+-ATPases and two distantly related sequences are located in each of the branches of Type I and Type V ATPases. As many as five Type IV ATPases are present, two of which are closely related. In addition, in the S. cerevisiae genome, three additional Type IID ATPases and two Type IIIB ATPases are found which are rather similar in sequence and each form small isoenzyme subfamilies.
P-type ATPases in Higher Organisms Not Yet Fully Sequenced
Analysis of expressed sequence tags (ESTs) provides a means to study the number and nature of genes in higher eukarya. Twenty-five eukaryotic sequences from all families of the phylogenetic tree were used to search the EST databases of Arabidopsis thaliana, C. elegans, and Homo sapiens for expressed P-type ATPases with similarity to already cloned pumps (Table 4). In all three organisms P-type ATPases were found that belong to the families of Type IB, IIA, IIB, IV, and V. There were, however, also marked differences between the single plant and the two animal (invertebrate and vertebrate, respectively) P-type ATPase related ESTs. Strikingly, in the EST database several related A. thaliana sequences showed similarity to Type IIIA ATPases (H+-pumps; Table 4), but no sequence showed similarity to Type IIC ATPases (Na+/K+- and H+/K+-pumps). On the contrary, several C. elegans and H. sapiens EST sequences were similar to Type IIC ATPases (Table 4), but no EST sequence from these organisms resembled Type IIIA ATPases.
Due to the low general quality of the sequences in the EST databases it was in some cases difficult to ascertain whether two sequences were actually identical or not. With these uncertainties in mind, there are, in the EST databases, between 18 to 27, 19 to 23 and 21 to 34 different P-type ATPase sequences represented from A. thaliana, C. elegans and H. sapiens, respectively (Table 4). A. thaliana appears to harbour at least 10 P-type H+-ATPases (Harper et al. 1994), five of which are represented by ESTs in the database (Table 4). This would suggest that far from all ATPases are present in the EST databases. A recent estimate suggests that about half of all Arabidopsis genes are represented by an EST (Rounsley et al. 1996).
Residues Determining Ion Specificity in Two Closely Related Families
Na+/K+ ATPases and H+/K+ ATPases (Type IIC ATPases; Fig. 6) are the two most closely related P-type ATPases having distinct ion specificities and most likely represent a recent development. We may assume that amino acids determining Na+ specificity are conserved in all Na+/K+-ATPases but may not be essential for the function of H+/K+-ATPases. Likewise amino acids determining H+ specificity are probably conserved in all H+/K+-ATPases. A comparison between all members of the two families (Table 1 and 2) reveal that amino acids at several positions are conserved in all Na+/K+-ATPases, but may vary in H+/K+-ATPases, and vice versa (Fig. 2). These amino acids are not randomly distributed in the sequences and only a few are overlapping with the universally conserved core regions (Fig. 2). Rather, they appear to be clustered in the trans-membrane segments (numbers 3, 6, 8, 9, and 10; Fig. 2) and in the stalk regions (Møller et al. 1996; Fig. 2) connecting the cytoplasmic domains with the membrane spanning segments. The criteria in this study for defining the various types of P-type ATPsaes has solely been the phylogenetic relationship between the conserved segments defined in Fig. 3.
DISCUSSION
This investigation shows P-type ATPases dividing into five major branches. This differs from earlier investigations dividing the P-type ATPase superfamily into two (Fagan and Saier 1994) or three (Lutsenko and Kaplan 1995) branches. The five major branches in the phylogenetic tree (Fig. 6) are Type I ATPases (heavy metal pumps), Type II ATPases (Ca2+-ATPases, Na+/K+-ATPases, and H+/K+-ATPases), Type III ATPases (H+ and Mg2+ pumps), Type IV ATPases (phospholipid pumps), and Type V ATPases (a group of pumps having no assigned substrate specificity). It should be noted, however, that this analysis is limited by the fact that the ion specificity of Type IIC ATPases has not been confirmed in all cases (Tables 1 and 2).
A Hitherto Unrecognized Family of P-type ATPases
A wealth of sequences in higher eukarya resembling Type V ATPases are found in the databases of ESTs (Table 4). This indicates that although poorly characterized, this branch of ATPases is large and ubiquitous among eukarya. An electrogenic chloride pump resembling a P-type ATPase has been characterized in the intestine of Aplysia californica (Gerencser and Purushotham 1996), anion-stimulated ATPase activities are found in several tissues and organisms (Gerencser 1996), and electrophysiological evidence points to an electrogenic chloride pump in the plasma membrane of Acetabularia (Gradmann et al. 1982). However, no P-type ATPase involved in the transport of any anion has so far been cloned. Since Type V ATPases form the only P-type ATPase family having no substrate specificities assigned to it, anions may be candidate substrates to be transported by them. Cloning of the respective genes will illuminate whether any of the P-type anion pumps belong to Type V ATPases.
Phospholipid Transporting P-type ATPases Form a Large Eukaryotic Branch of P-type ATPases
Only one Type IV ATPase (seq. 14; Fig. 6) has been characterized at the biochemical level (Auland et al. 1994). This Bos taurus pump transports aminophospholipids such as phosphatidylserine and phosphatidylethanolamine (Auland et al. 1994). Five Type IV ATPases are found in the S. cerevisiae genome (Tables 1 and 3 ). Deletion of one of these sequences (seq. 15; Fig. 6) abolishes trans-membrane phosphatidylserine transport, suggesting that the S. cerevisiae and the B. taurus genes have similar functions (Tang et al. 1996). This suggests that trans-membrane transport of various phospholipids, which would contribute to establishing lipid bilayer asymmetry, is a general feature of the poorly characterized branch of Type IV ATPases. Type IV ATPases are not found in bacteria and archaea but are represented abundantly in eukaryotes as evident from their appearance in the EST database (Table 4).
Type IIA and IIB Ca2+-ATPases are Both Present in a Variety of Membranes
Type IIA Ca2+-ATPases are mainly present in the sarcoplasmic and endoplasmic reticulum but in addition includes a plant pump (seq. 150; Fig. 6) that seems to be present in both the vacuolar membrane and the plasma membrane (Ferrol and Bennett 1996). Type IIB Ca2+-ATPases are primarily found in the plasma membrane but this family also includes pumps (seq. 119, 128, and 129; Fig. 6) present in the vacuolar membrane (Cunningham and Fink 1994; Moniakis et al. 1995; Malmström et al. 1997). Since members of both families can be present in a variety of membranes it does not seem appropriate to name these families according to intracellular localization, i.e. SERCA pumps for sarco-endo-plasmatic reticulum Ca2+-ATPases and PMCA pumps for plasma membrane Ca2+-ATPases, but rather Type IIA and Type IIB Ca2+-ATPases, respectively, according to their phylogenetic relationship.
Complementary Function of Type III and Type IIC ATPases
H+ (Type IIIA)- and Na+/K+ (Type IIC)- transporting P-type ATPases form large eukaryotic families. Analysis of EST databases (Table 4) suggests that among plants, fungi and animals, plasma membrane H+-ATPases are specific for fungi and plants, and, on the other hand, that plasma membrane Na+/K+-ATPases are distinguishing features of animal cells. Secondary active transport in fungi and plants is energized by H+ gradients, whereas in animals Na+ gradients are used as an energy source for such transport systems (Skulachev 1994). Therefore, Type IIIA and Type IIC pumps seem to serve complementary functions in the various classes of eukarya.
Fungi apparently form an intermediate group harbouring both H+- and Na+-pumps, the latter in a distinct branch (Type IID ATPases) separate from that of animal Na+/K+- ATPases. However, it has not been conclusively demonstrated that Type IID ATPases transport Na+. The ENA genes (ENA1; seq. 94) were suggested to be Na+ pumps based on genetic evidence demonstrating that deletion of the genes confer Na+ sensitivity to S. cerevisiae (Haro et al. 1991; Garciadeblas et al. 1993). Nevertheless, disruption of the related CTA3 gene (seq. 93) in S. pombe leads to higher levels of cytosolic free Ca2+ (Ghislain et al 1990) in addition to reduced Ca2+ in intracellular organelles (Halachmi et al. 1992), suggesting that this pump is involved in removing Ca2+ from the cytosol to intracellular stores. Since Na+ tolerance in S. cerevisiae is regulated by Ca2+ (Mendoza et al., 1994; Hirata et al., 1995) it remains a theoretical possibility that the ENA genes encode Ca2+-ATPases essential for regulating intracellular Ca2+ during Na+ adaptation. A biochemical characterization of Type IID ATPases is needed in order to clarify this question.
It is uncertain whether P-type H+- or Na+-ATPases are found in bacterial species. H+-ATPase activity sensitive to the P-type ATPase inhibitor vanadate has been characterized in a cyanobacterium (Fresneau et al. 1993) and at least the solitary P-type ATPase in the archaeon M. jannaschii is very similar to H+-ATPases (Fig. 6). A single Synechocystis PCC6803 sequence (seq. 96; Fig. 6; Table 1) has some similarity to Type IIC ATPases although it may also be an unusual Type IIA Ca2+-ATPase.
KdpB ATPases May Represent an Ancient Group of P-type ATPases
No bacterial P-type ATPases have been cloned that group with Type IIB, Type IV, and Type V ATPases. Therefore, these ATPases most likely represent more recent evolutionary achievements. Most of the bacterial P-type ATPases are found in the branch of Type IB ATPases, in the related family of KdpB (Type IA) ATPases, and in the branch of Type IIA Ca2+-ATPases (Fig. 6). KdpB ATPases have an unusual trans-membrane organization compared to other P-type ATPases in having a reduced number of trans-membrane spans (Lutsenko and Kaplan 1995). In addition, unlike all other P-type ATPases, these ATPases have three subunits (Hesse et al 1984). KdpB is the catalytic subunit, whereas KdpA is involved in the binding of K+, and KdpC seems to stabilize the complex between KdpB and KdpA (Buurman et al. 1995). Fusion of genes occurred during the evolution of bacteria (Miozzari and Yanofsky 1979) and KdpB ATPases may represent ancestral P-type ATPase which acquired new trans-membrane helices and the ability to bind the transported substrate, for example, by fusion of the catalytic subunit with substrate binding proteins such as KdpA encoded within the same operon. The subunit of the heterodimeric Na+/K+- and H+/K+-ATPases (Type IIC), which plays a role in stabilization of the catalytic  subunit (Møller et al. 1996), may be a reminiscent of the KdpC subunit whereas at least H+- (Type IIIA) and Ca2+- (Type IIA and Type IIB) ATPases seem to have eliminated this subunit.
subunit (Møller et al. 1996), may be a reminiscent of the KdpC subunit whereas at least H+- (Type IIIA) and Ca2+- (Type IIA and Type IIB) ATPases seem to have eliminated this subunit.
P-type ATPases Group in a Phylogenetic Tree According to Substrate Specificity
The substrate specificities of most P-type ATPases cloned so far are not known. Obtaining this information requires, for example, demonstration of amino acid identity to biochemically well-characterized proteins or expression of the genes in heterologous systems followed by biochemical characterization of the gene products. However, in each of the described branches of ATPases presented in Fig. 6 it is noteworthy that only single ion specificities have been found for those proteins in the branch that have been characterized with respect to function. The only exception to this rule so far is Type IID ATPases (see above).
The various P-type ATPases present in single organisms are often more related to P-type ATPases in evolutionarily distant species, than they are to endogenous P-type ATPases in other branches. For example, plasma membrane H+-ATPases of S. cerevisiae (seq. 86 and 87; Fig. 6) and the plant A. thaliana (seq. 68; Fig. 6) show high similarity to each other and are analogous enzymes with respect to function (Palmgren and Christensen 1993; Supply et al. 1993). On the contrary, all the remaining 14 P-type ATPases in S. cerevisiae (Table 1 and 2) have significantly weaker similarity to the H+-ATPase sequences. The H+-ATPase sequences are closely related to the Type IIIA P-type ATPase of the archaeon M. jannaschii (60% sequence identity within core sequences). The divergence between the M. jannaschii P-type ATPase and the H+-ATPases most likely took place about one and a half billion years ago, which is an estimated date for the divergence of eukarya and archaea (Doolittle et al. 1996). In contrast, there is less than 25% identity within core sequences between, for example, any Type IV and any Type I ATPase. Assuming that two diverging protein sequences change in a more or less stochastic manner consistent with an exponential decay (Doolittle 1995), the duplication giving rise to Type IV and Type V ATPases thus occurred several billion years ago and clearly preceded the divergence of eukarya and bacteria (or life for that matter). Still, we do not find any genes with detectable similarity to Type IV and Type V ATPases in the completed genomes of four bacteria and one archaeon. Although in theory it is possible that these organisms have eliminated, for example, Type IV and Type V ATPases from their genomes relative recently, the complete absence of these ATPases in prokaryotes argues against the proposition that Type IV ATPases represent an ancient group of P-type pumps (Tang et al. 1996). A plausible explanation would be that P-type ATPases have not evolved at a constant rate, and that Type IV and Type V ATPases are relatively recent evolutionary achievements that evolved more rapidly than other P-type ATPases.
Several Amino Acid Residues May be Involved in Determining Ion Specificity of P-type ATPases
It is generally believed that with ion pumps changing, only a few or a single amino acid residue may be sufficient in order to develop a new ion specificity (Sasaki et al. 1995; Zhang and Fillingame 1995). If a single or very few residues are sufficient determinants of ion specificity in P-type ATPases, we would expect to find many examples of closely related pumps having different ion specificity. However, this is not the case. Several amino acid residues may be required in determining the shape and specificity of the ion binding site(s) near or inside the proposed ion channel (Møller et al. 1996) of P-type ATPases. It is thus possible that several, if not all, of the residues characteristic for each of the closely related Na+/K+- and H+/K+-ATPase families (indicated by boxes in Fig. 2) are required in order to obtain Na+ and H+ specificities, respectively. In accordance with this observation, mutagenesis of the conserved Na+/K+-ATPase glutamate-961 into glutamine (which is present in all H+/K+-ATPases), aspartate, alanine, or leucine slightly reduces the Na+ affinity of the ATPase but does not abolish the transporting capabilities of the enzyme (Van Huysse and Lingrel 1993). Substitutions of serine-782 (present in all Na+/K+-ATPases but not in H+/K+-ATPases) does not result in any detectable changes in Na+ affinity (Argüello and Lingrel 1995). A concerted action between several residues as a requirement for obtaining absolute ion specificity would explain the large divergence in single species between P-type ATPases having different ion specificity.
In conclusion, P-type ATPases cluster in the phylogenetic tree according to substrate specificity despite the evolutionary distance between the parental species. This suggests that, firstly, within a given substrate specificity of P-type ATPases there are limits for evolutionary changes. Secondly, in order for a change in substrate specificity to occur, any P-type ATPase has to be released from a structural constraint that is subsequently followed by a dramatic change in primary structure. Actins are among the slowest-changing proteins known probably because actin evolution is limited by structural constraints that has to do with the large number of protein-protein interactions they must preserve (Doolittle 1995). Although actins evolve extremely slowly, they show little sequence identity to their closest relatives, eukaryotic centractins and bacterial ftsA proteins. This suggests abrupt changes in the rate of sequence evolution immediately following the divergence from common ancestors (Doolittle 1995). In this respect, the evolution of actins and the P-type ATPase superfamily resemble each other.
Acknowledgements
This work was supported by the European Union Biotechnology Program BIO4-CT-96-0775. We are indebted to Jeff Harper for useful suggestions and stimulating discussions. We would like to thank Hans Ullitz Møller and Bo Mikkelsen at Biobase for assistance with computational problems, Henrik Nielsen at the Centre for Biological Sequence Analysis for help with the helix predictions and Lone Baunsgaard, Thomas Jahn, Kirk Schnorr, and Kees Venema for critically reading the manuscript.
References
Argüello JM, Lingrel JB (1995) Substitutions of serine 775 in the subunit of the Na,K-ATPase selectively disrupt K+ high affinity activation without affecting Na+ interaction. J Biol Chem 270:22764-22771
Auland ME, Roufogalis BD, Devaux PF, Zachowski A (1994) A Reconstitution of ATP-dependent aminophospholipid translocation in proteoliposomes. Proc Natl Acad Sci USA 91:10938-10942
Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW (1993) The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet 5:327-337
Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, FitzGerald LM, Clayton RA, Gocayne JD, Kerlavage AR, Dougherty BA, Tomb JF, Adams MD, Reich CI, Overbeek R, Kirkness EF, Weinstock KG, Merrick JM, Glodek A, Scott JL, Geoghagen NSM, Weidman JF, Fuhrmann JL, Nguyen D, Utterback TR, Kelley JM, Peterson JD, Sadow PW, Hanna MC, Cotton MD, Roberts KM, Hurst MA, Kaine BP, Borodovsky M, Klenk H-P, Fraser CM, Smith HO, Woese CR, Venter JC (1996) Complete genome sequence of the methanogenic Archaeon, Methanococcus jannaschii. Science 273:1058-1073
Buurman ET, Kim KT, Epstein W (1995) Genetic evidence for two sequentially occupied K+ binding sites in the Kdp transport ATPase. J Biol Chem 270:6678-6685
Cunningham KW, Fink GR (1994) Calcineurin-dependent growth control in Saccharomyces cerevisiae mutants lacking PMC1, a homolog of plasma membrane Ca2+ ATPases. J Cell Biol 124:351-363
Doolittle RF (1995) The origins and evolution of eukaryotic proteins. Philos Trans R Soc Lond B Biol Sci 349:235-240
Doolittle RF, Feng DF, Tsang S, Cho G, Little E (1996) Determining divergence times of the major kingdoms of living organisms with a protein clock. Science 271:470-477
Fagan MJ, Saier MH Jr (1994) ATPases of eukaryotes and bacteria: Sequence analyses and construction of phylogenetic trees. J Mol Evol 38:57-99
Felsenstein J (1989) PHYLIP - phylogeny inference package (Version 32). Cladistics 5:164-166
Ferrol N, Bennett AB (1996) A single gene may encode differentially localized Ca2+-ATPases in tomato. Plant Cell 8:1159-1169
Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, McKenney K, Sutton G, Fitzhugh W, Fields C, Gocayne JD, Scott J, Shirley R, Liu LI, Glodek A, Kelley JM, Weidman JF, Phillips CA , Spriggs T, Hedblom E, Cotton MD, Utterback TR, Hanna MC, Nguyen DT, Saudek DM, Brandon RC, Fine LD, Fritchman JL, Fuhrmann JL, Geoghagen NSM, Gnehm CL, McDonald LA, Small KV, Fraser CM, Smith HO, Venter JC (1995) Whole-genome random sequencing and assembly of Haemophilus influenzae. Science 269:496-512
Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, Fleischmann RD, Bult CJ, Kerlavage AR, Sutton G, Kelley JM, Fritchman JL, Weidman JF, Small KV, Sandusky M, Fuhrmann J, Nguyen D, Utterback TR, Saudek DM, Philips CA, Merrick JM, Tomb J-F, Dougherty BA, Bott KF, Hu P-C, Lucier TS, Peterson SN, Smith HO, Hutchison CA, III, Venter JC (1995) The minimal gene complement of Mycoplasma genitalium. Science 270:397-403
Fresneau C, Riviere ME, Arrio B (1993) Characterization of the plasmalemma ATPase from the cyanobacteria Synechococcus PCC 6311 and PCC 7942. Arch Biochem Biophys 306:254-260
Garciadeblas B, Rubio F, Quintero FJ, Banuelos MA, Haro R, Rodriguez-Navarro A (1993) Differential expression of two genes encoding isoforms of the ATPase involved in sodium efflux in Saccharomyces cerevisiae. Mol Gen Genet 238:363-368
Gerencser GA (1996) The chloride pump: a Cl--translocating P-type ATPase. Crit Rev Biochem Mol Biol 31:303-337
Gerencser GA, Purushotham KR (1996) Reconstituted Cl- pump protein: a novel ion(Cl-)-motive ATPase. J Bioenerg Biomembr 28:459-469
Ghislain M, Goffeau A, Halachmi D, Eilam Y (1990) Calcium homeostasis and transport are affected by disruption of cta3, a novel gene encoding Ca2+-ATPase in Schizosaccharomyces pombe. J Biol Chem 265:18400-18407
Goffeau A. et al. (1997) The yeast genome directory. Nature (supplement) 387:1-105
Gradmann D, Tittor J, Goldfarb V (1982) Electrogenic Cl- pump in Acetabularia. Philos Trans R Soc Lond B 299:447-457
Halachmi D, Ghislain M, Eilam Y (1992) An intracellular ATP-dependent calcium pump within the yeast Schizosaccharomyces pombe, encoded by the gene cta3. Eur J Biochem 207:1003-1008
Haro R, Garciadeblas B, Rodriguez-Navarro A (1991) A novel P-type ATPase from yeast involved in sodium transport. FEBS Lett 291:189-91
Harper JF, Manney L, Sussman MR (1994) The plasma membrane H+-ATPase gene family in Arabidopsis: genomic sequence of AHA10 which is expressed primarily in developing seeds. Mol Gen Genet 244:572-587
Hesse JE, Wieczorek L, Altendorf K, Reicin AS, Dorus E, Epstein W (1984) Sequence homology between two membrane transport ATPases, the Kdp-ATPase of Escherichia coli and the Ca2+-ATPase of sarcoplasmic reticulum. Proc Natl Acad Sci USA 81:4746-4750
Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, Herrmann R (1996) Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res 24:4420-4449
Hirata D, Harada S I, Namba H, Miyakawa T (1995) Adaptation to high-salt stress in Saccharomyces cerevisiae is regulated by Ca2+/calmodulin-dependent phosphoprotein phosphatase (calcineurin) and cAMP-dependent protein kinase. Mol Gen Genet 249:257-264
Lingrel JB, Kuntzweiler T (1994) Na+,K+-ATPase. J Biol Chem 269:19659-19662
Lutsenko S, Kaplan JH (1995) Organization of P-type ATPase: Significance of structural diversity. Biochemistry 34:15607-15612
Malmström S, Askerlund P, Palmgren MG (1997) A calmodulin-stimulated Ca2+-ATPase from plant vacuolar membranes with a putative regulatory domain at its N-terminus. FEBS Lett 400:324-328
Mendoza I, Rubio F, Rodriguez-Navarro A, Pardo J M (1994) The protein phosphatase calcineurin is essential for NaCl tolerance of Saccharomyces cerevisiae. J Biol Chem 269:8792-8796
Miozzari GF, Yanofsky C (1979) Gene fusion during the evolution of the tryptophan operon in Enterobacteriaceae. Nature 277:486-489
Moniakis J, Coukell MB, Forer A (1995) Molecular cloning of an intracellular P-type ATPase from Dictyostelium that is up-regulated in calcium-adapted cells. J Biol Chem 270:28276-28281
Møller JV, Juul B, Le Maire M (1996) Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim Biophys Acta 1286:1-51
O'Brien C (1997) Entire E. coli genome sequenced - at last. Nature 385:472
Olsen GJ, Woese CR, Overbeek R (1994) The winds of (evolutionary) change: Breathing new life into microbiology. J Bacteriol 176:1-6
Palmgren MG, Christensen G (1993) Complementation in situ of the yeast plasma membrane H+-ATPase gene pma1 by an H+-ATPase gene from a heterologous species. FEBS Lett 317:216-222
Program Manual for the Wisconsin Package, Version 8 September 1994 Genetics Computer Group, 575 Science Drive, Madison, Wisconsin, USA 53711 (1994)
Rounsley SD, Glodek A, Sutton G, Adams MD, Somerville CR, Venter JC, Kerlavage AR (1996) The construction of Arabidopsis expressed sequence tag assemblies A new resource to facilitate gene identification. Plant Physiol 112:1177-1183
Sasaki J, Brown LS, Chon YS, Kandori H, Maeda A, Needleman R, Lanyi JK (1995) Conversion of bacteriorhodopsin into a chloride ion pump. Science 269:73-75
Serrano R (1988) Structure and function of proton translocating ATPase in plasma membranes of plants and fungi. Biochim Biophys Acta 947:1-28
Skulachev VP (1994) Bioenergetics: the evolution of molecular mechanisms and the development of bioenergetic concepts. Antonie van Leeuwenhoek 65:271-284
Supply P, Wach A, Thines-Sempoux D, Goffeau A (1993) Proliferation of intracellular structures upon overexpression of the PMA2 ATPase in Saccharomyces cerevisiae. J Biol Chem 268:19744-19752
Tang X, Halleck MS, Schlegel RA, Williamson P (1996) A subfamily of P-type ATPases with aminophospholipid transporting activity. Science 272:1495-1497
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673-4680
Van Huysse JW, Lingrel JB (1993) Nonpolar amino acid substitutions of potential cation binding residues Glu-955 and Glu-956 of the rat 1 isoform of Na+, K+-ATPase. Cell Mol Biol Res 39:497-507
Von Heijne G (1992) Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol 225:487-494
Zhang Y, Fillingame RH (1995) Changing the ion binding specificity of the Escherichia coli H+-transporting ATP synthase by directed mutagenesis of subunit c. J Biol Chem 270:87-93
FIGURES
Fig. 1.
Graphic presentation of various P-type ATPase sequences.
In the upper part of the protein the conserved segments are shown (in white), while in the lower part, predicted heavy metal binding sites are shown (in grey) together with predicted trans membrane segments (in white). The proteins are aligned at the conserved segment A (shown in white) common to all P-type ATPases. The length of each ATPase is given at the end. At the bottom an example shows how the core sequences were extracted. These were subsequently used for the phylogenetic analysis. The numbers to the left correspond to the numbers used in Table 1. The prediction of transmembrane segments was performed according to von Heijne (1992) but has only in a few cases been verified by other methods.

Fig. 2.
A model of Na+/K+-ATPase showing the localization of core segments relative to transmembrane segments and conserved amino acid positions in Na+/K+- and H+/K+-ATPases.
Core segments are shown in black. The sequences of 26 cloned Na+/K+-ATPases and 11 cloned H+/K+-ATPases were aligned and amino acid identities between and within the two groups were determined. The empty circles represent amino acid positions which are variant. The amino acids shown in circles are identical among all Na+/K+- and H+/K+-ATPases. The squares show positions where amino acids are identical within one of the two groups with the Na+/K+-ATPases shown first. A dash is shown in the squares if the position is only identical in one of the two groups and the same amino acid or a conservative substitution is not present at that position in more than 20% of the sequences of the other group. A dot is shown if all sequences in the group have a gap at this position compared to the other group. The amino acid stretches where several squared positions are present are placed in frames. The membrane topology and numbering of residues correspond to the human Na+/K+-ATPase 1 isoform (seq. 104 in Table 1). The prediction of transmembrane segments was performed according to von Heijne (1992) and resembles with respect to the number of transmembrane spans the prediction by Lingrel and Kuntzweiler (1994).
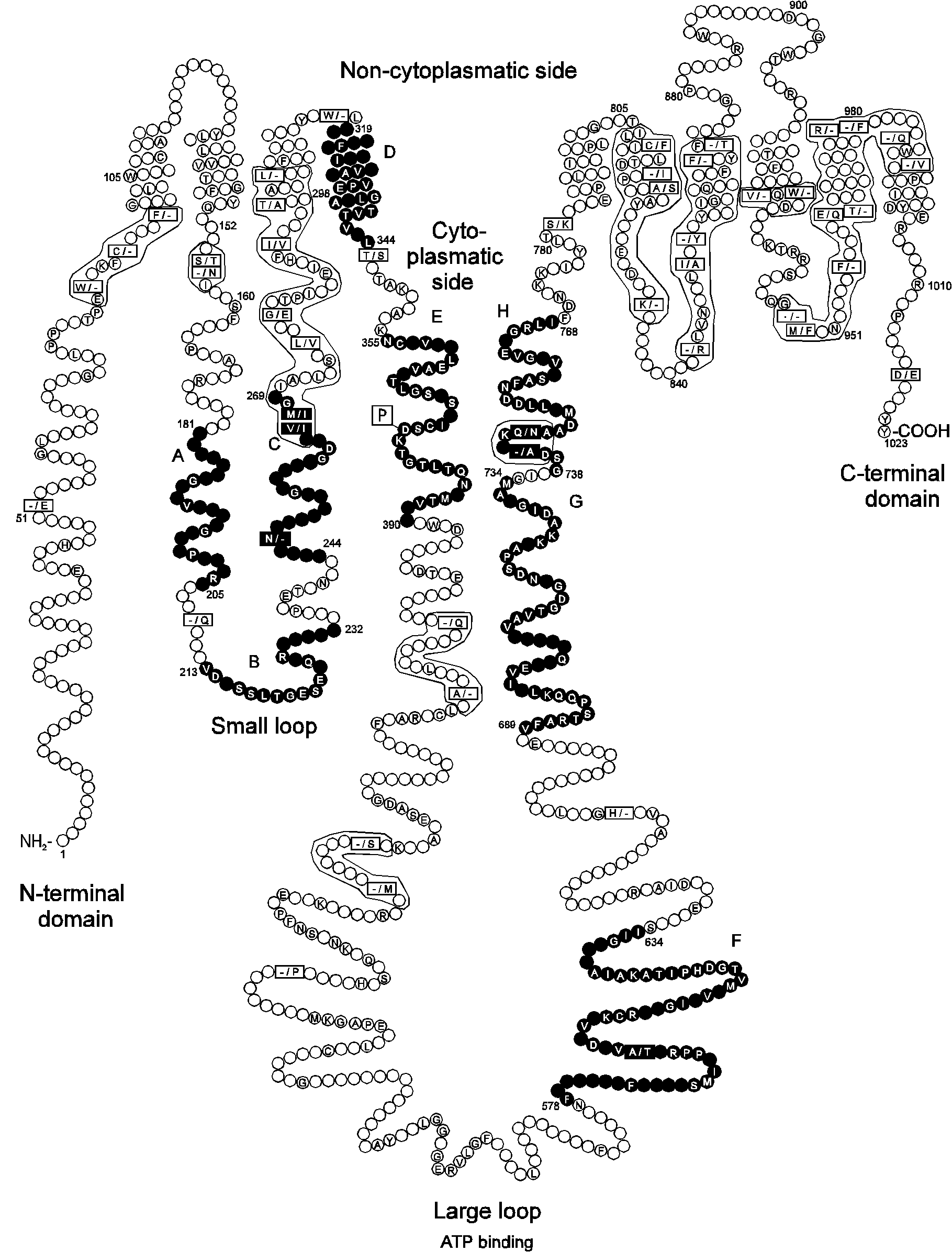
Fig. 3.
Alignment of conserved segments of the 20 P-type ATPases shown in Fig. 1.
Repetitive lowercase letters on top indicate name of conserved segment. Amino acids present in at least 135 out of 159 analyzed sequences (Table 1) are in bold. Before and after each of the conserved segments the position in the original sequence is given. Numbers at top indicate the position of amino acids when conserved segments are arranged in a linear core sequence. Sequence numbers correspond to the numbers used in Table 1.
1 13 25 26 45 46 71 72
Type Segments aaaaaaaaaaaaa aaaaaaaaaaaa bbbbbbbbbbbbbbbbbbbb cccccccccccccccccccccccccc ddddddddddddddd
IA Seq. 20 122 PADQLRKGDIVLV EAGDIIPCDGEV 146..153 VDESAITGESAPVIRESGGD 172..177 TGGTRILSDWLVIECSVNPGETFLDR 202..252 VTVLVALLVCLIPTT
IB Seq. 27 153 PISELKMGDQVLV KPGELVPTDGLV 177..184 LNQASITGESMPVEKAIGDE 203..205 FAGTINGNGVLRLKIHQPPESSLIQR 230..283 IYRALIFLVVASPCA
IB Seq. 41 262 PASELKKRQRFVT RPGETIAADGVV 286..293 IDMSAMTGEAKPVRAYPAAS 312..314 VGGTVVMDGRLVIEATAVGADTQFAA 339..391 FSVTLGVLVIACPCA
IB Seq. 48 427 PIELLQVNDIVEI KPGMKIPADGII 451..458 IDESLMTGESILVPKKTGFP 477..479 IAGSVNGPGHFYFRTTTVGEETKLAN 504..572 LQTATSVVIVACPCA
IB Seq. 49 838 DVELVQRGDIIKV VPGGKFPVDGRV 862..869 VDESLITGEAMPVAKKPGST 888..890 IAGSINQNGSLLICATHVGADTTLSQ 915..989 FQASITVLCIACPCS
IIA Seq. 154 149 PVAGLVPGDLILL EAGDQVPADARL 173..181 VKESALTGEAEAVQKLADQQ 200..215 FQGTEVLQGRGQALVYATGMNTELGR 240..292 LSVGLSMAVAIVPEG
IIA Seq. 141 141 KAKDIVPGDIVEI AVGDKVPADIRL 165..175 VDQSILTGESVSVIKHTDPV 194..209 FSGTNIAAGKAMGVVVATGVNTEIGK 234..296 FKIAVALAVAAIPEG
IIB Seq. 123 202 PVAEIVVGDIAQV KYGDLLPADGIL 226..234 IDESSLTGESDHVKKSLDKD 253..257 LSGTHVMEGSGRMVVTAVGVNSQTGI 282..410 FIIGITVLVVAVPEG
IIC Seq. 104 181 NAEEVVVGDLVEV KGGDRIPADLRI 205..213 VDNSSLTGESEPQTRSPDFT 232..244 FFSTNCVEGTARGIVVYTGDRTVMGR 269..321 VIFLIGIIVANVPEG
IIIA Seq. 61 127 PAKELVPGDVVRI RIGDIVPADIIL 151..159 VDESALTGESLPVEKKIGDI 178..180 YSGSIVKKGEMTGIVKATGLNTYFGK 205..256 AQFALVLAVSAIPAA
IIIA Seq. 86 192 PANEVVPGDILQL EDGTVIPTDGRI 216..225 IDQSAITGESLAVDKHYGDQ 244..246 FSSSTVKRGEGFMVVTATGDNTFVGR 271..323 LRYTLGITIIGVPVG
IIIA Seq. 68 144 EAAILVPGDIVSI KLGDIIPADARL 168..176 VDQSALTGESLPVTKHPGQE 195..197 FSGSTCKQGEIEAVVIATGVHTFFGK 222..274 IDNLLVLLIGGIPIA
IIIB Seq. 59 175 PIDQLVPGDIIKL AAGDMIPADLRI 199..207 VAQASLTGESLPVEKAATTR 226..241 FMGTTVVSGTAQAMVIATGANTWFGQ 266..318 ALFALSVAVGLTPEM
IV Seq. 9 266 PSKDLKVGDLIKV HKGDRIPADLVL 290..302 IKTDQLDGETDWKLRVACPL 321..364 VDNTLWANTVLASSGFCIACVVYTGR 389..444 ILRYLILFSTIIPVS
IV Seq. 14 147 HWEKVNVGDIVII KGKEYIPADTVL 171..183 IETSNLDGETNLKIRQGLPA 202..247 ADQILLRGAQLRNTQWVHGIVVYTGH 272..344 FLTFIILFNNLIPIS
IV Seq. 11 164 EWRYILVGDFVHI SNNQDVPADIIL 188..200 IETCNLDGETSLKQRMVPAK 219..267 KENMLLRGSRIKNTTFVEGIVVYAGH 292..367 IGAFFINYQLLVPIS
V Seq. 4 112 SSSELVPGDIYEI KNNMTIPCDTII 136..143 MSEHMLTGESVPIHKERLPF 162..363 IKYNNKEENRILGLVIKTGFITTKGK 388..440 IIKCLDIITDAIPPA
V Seq. 2 258 GSDQLVPGDILLI+ PHGCLMQCDSVL 283..290 VNESVLTGESVPITKVALTD 308..332 LQTRFYRGKKVKAIVLRTAYSTLKGQ 357..409 IVRSLDIITITVPPA
V Seq. 1 565 SSSELVPGDIYEV++ PNITILPCDSIL 591..598 VNESMLTGESVPVSKFPATE 617..649 RARIAPGQTAALAMVVRTGFSTTKGS 672..724 ILRALDIITIVVPPA
V Seq. 6 268 QTNELLPMDLVSI+++AEESAIPCDLIL 295..302 VNEAMLSGESTPLLKESIKL 321..355 SDIPPPPDGGALAIVTKTGFETSQGS 380..432 ILDCILIITSVVPPE
95 96 131 132 166
Type Segments ddddddddd eeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeee fffffffffffffffffffffffffffffffffff ffffffffffffffffff
IA Seq. 20 IGGLLSAIG 275...286 NVIATSGRAVEAAGDVDVLLLDKTGTITLGNRQASE 321...432 VVVEGSRVLGVIALKDIVKGGIKEAFAQLRKMGIK TVMITGDNRLTAAAIAA
IB Seq. 27 LMASIMPAL 306...317 GILFKNGAQLERIGRVRVIAFDKTGTLTTGKPEVVN 352...451 WVAYAGEILGLIAVADTVRPTAAQAIARLKRLGIE+IVMLTGDNSRTAHSIAQQ
IB Seq. 41 LGLATPTAM 414...425 GIFIKGYRALETIRSIDTVVFDKTGTLTVGQLAVST 460...560 FVSVDGVVRAALTIADTLKDSAAAAVAALRSRGLR TILLTGDNRAAADAVAAQ
IB Seq. 48 LGLATPTAI 595...606 GVLIKGGEVLEKFNSITTFVFDKTGTLTTGFMVVKK 641...748 YVSVNGHVFGLFEINDEVKHDSYATVQYLQRNGYE TYMITGDNNSAAKRVARE
IB Seq. 49 LGLATPTAV 1012..1023 GILIKGGEPLEMAHKVKVVVFDKTGTITHGTPVVNQ 1058..1215 LVAVDDELCGLIAIADTVKPEAELAIHILKSMGLE VVLMTGDNSKTARSIASQ
IIA Seq. 154 LPAVITVAL 315...326 ESLIRRLPAVETLGSVTTICSDKTGTLTQNKMVVQQ 361...548 DAETDLTWLGLMGQIDAPRPEVREAVQRCRQAGIR TLMITGDHPLTAQAIARD
IIA Seq. 141 LPAVITTCL 319...330 NAIVRSLPSVETLGCTSVICSDKTGTLTTNQMSVCR 365...585 KYETNLTFVGCVGMLDPPRIEVASSVKLCRQAGIR VIMITGDNKGTAVAICRR
IIB Seq. 123 LPLAVTISL 433...444 NNLVRHLDACETMGNATAICSDKTGTLTMNRMTVVQ 479...657 EILTELTCIAVVGIEDPVRPEVPDAIAKCKQAGIT VRMVTGDNINTARAIATK
IIC Seq. 104 LLATVTVCL 344...355 NCLVKNLEAVETLGSTSTICSDKTGTLTQNRMTVAH 390...578 FPIDNLCFVGLISMIDPPRAAVPDAVGKCRSAGIK VIMVTGDHPITAKAIAKG
IIIA Seq. 61 MPAVLSITM 279...290 DAIVKKLVAIEELAGVDILCSDKTGTLTKNQLVCGE 325...442 YKNGRWHFAGIIPLYDPPREDAPLAVKKIKELGVI IKMVTGDHVAIAKNIARM
IIIA Seq. 86 LPAVVTTTM 346...357 QAIVQKLSAIESLAGVEILCSDKTGTLTKNKLSLHE 392...519 RGEGHWEILGVMPCMDPPRDDTAQTVSEARHLGLR VKMLTGDAVGIAKETCRQ
IIIA Seq. 68 MPTVLSVTM 297...308 GAITKRMTAIEEMAGMDVLCSDKTGTLTLNKLSVDK 343...472 SPGAPWEFVGLLPLFDPPRHDSAETIRRALNLGVN VKMITGDQLAIGKETGRR
IIIB Seq. 59 LPMIVTSTL 341...352 KVIVKHLDAIQNFGAMDILCTDKTGTLTQDKIVLEN 387...530 ADESDLILEGYIAFLDPPKETTAPALKALKASGIT VKILTGDSELVAAKVCHE
IV Seq. 9 LRVNLDLAK 467...482 ETIVRTSTIPEDLGRIEYLLSDKTGTLTQNDMQLKK 517...735 YLEHDLELLGLTGVEDKLQKDVKSSIELLRNAGIK IWMLTGDKVETARCVSIS
IV Seq. 14 LLVTLEVVK 367...388 AAMARTSNLNVELGQVKYIFSDKTGTLTCNVMQFKK 423...630 LIEKNLQLLGATAIEDKLQDQVPETIETLMKADIK IWILTGDKQETAINIGHS
IV Seq. 11 LYITVEIIK 390...411 AIDCRSLSIPEELGTVTHVLSDKTGTLTENMMIFRN 446...798 ELETNLKLSGVTGIEDRLQDGVPDTLRALRDAGIQ VWVLTGDKLETAQNIATS
V Seq. 4 LPTTLTVGI 463...475 SISCLCPHKINIAGQINTMVFDKTGTLTENNLQFIG 510..1004 FVESNLHFLGFLIFTNNMKKNAPDIIHNLQTSGCQ CIMSTGDNVLTSIHVAKK
V Seq. 2 LPAAMSVGI 433...444 EIFCISPSTINTCGAINVVCFDKTGTLTEDGLDFHV 479...680 AVECDLEMLGLIVMENRVKPVTLGVINQLNRANIR TVMVTGDNLLTGLSVARE
V Seq. 1 LPATLTIGT 749...760 GIFCISPTRLNISGKIDVMCFDKTGTLTEDGLDVLG 795..1014 EVESNLEFLGFIIFQNKLKKETSETLKSLQDANIR TIMCTGDNILTAISVGRE
V Seq. 6 LPMELTMAV 455...466 YVYCTEPFRIPFAGRIDVCCFDKTGTLTGEDLVFEG 501...658 DVESELTFNGFLIFHCPLKDDAIETIKMLNESSHR SIMITGDNPLTAVHVAKE
188 189 207 234 235 265
Type Segments ffff ggggggggggggggggggg ggggggggggggggggggggggggggg hhhh hhhhhhhhhhhhhhhhhhhhhhhhhhh Total
IA Seq. 20 AGVD 488...490 FLAEATPEAKLALIRQYQA EGRLVAMTGDGTNDAPALAQADVAVAM 535...538 GTQA AKEAGNMVDLDSNPTKLIEVVHIGKQM 568...682
IB Seq. 27 VGVN 508...509 VYAELLPEDKVDVIRQLQK+ QYQSVAMVGDGINDAPALAQASVGIAM 555...559 GSDV ALETADIVLMADRLERLEHAIRLGRRA 589...642
IB Seq. 41 VGID 616...618 AVADMLPEGKVDVIQRLRE EGHTVAMVGDGINDGPALVGADLGLAI 663...666 GTDV ALGAADIILVRDDLNTVPQALDLARAT 696...752
IB Seq. 48 VGIS 804...808 VYSDVSPTGKCDLVKKIQD++ GNNKVAVVGDGINDAPALALSDLGIAI 855...858 GTEI AIEAADIVILCGNDLNTNSLRGLANAI 888..1004
IB Seq. 49 VGIT 1271..1273 VFAEVLPSHKVAKVKQLQE EGKRVAMVGDGINDSPALAMANVGIAI 1318..1321 GTDV AIEAADVVLIRNDLLDVVASIDLSRKT 1351..1500
IIA Seq. 154 LGIT 604...633 VYARVAPEHKLRIVESLQR QGEFVAMTGDGVNDAPALKQANIGVAM 678...682 GTDV SKEASDMVLLDDNFATIVAAVEEGRIV 712...926
IIA Seq. 141 IGIF 641...674 CFARVEPSHKSKIVEFLQS FDEITAMTGDGVNDAPALKKAEIGIAM 719...722 GTAV AKTASEMVLADDNFSTIVAAVEEGRAI 752..1042
IIB Seq. 123 CGIL 713...752 VLARSSPTDKHTLVKGIID+++++HRQVVAVTGDGTNDGPALKKADVGFAM 802...806 GTDV AKEASDIILTDDNFTSIVKAVMWGRNV 836..1205
IIC Seq. 104 VGII 634...689 VFARTSPQQKLIIVEGCQR QGAIVAVTGDGVNDSPALKKADIGVAM 734...738 GSDV SKQAADMILLDDNFASIVTGVEEGRLI 768..1023
IIIA Seq. 61 LGIG 498...530 GFAEVFPEHKYKIVDSLQK RGHLVAMTGDGVNDAPALKKADCGIAV 575...578 ATDA ARAAADIVLLSPGISVIVDAIQEARRI 608.. 805
IIIA Seq. 86 LGLG 575...606 GFAEVFPQHKYRVVEILQN RGYLVAMTGDGVNDAPSLKKADTGIAV 651...654 ATDA ARSAADIVFLAPGLSAIIDALKTSRQI 684...919
IIIA Seq. 68 LGMG 528...560 GFAGVFPEHKYEIVKKLQE RKHIVGMTGDGVNDAPALKKADIGIAV 605...608 ATDA ARGASDIVLTEPGLSVIISAVLTSRAI 638...948
IIIB Seq. 59 VGLD 586...613 LFARLTPMHKERIVTLLKR EGHVVGFMGDGINDAPALRAADIGISV 658...661 AVDI AREAADIILLEKSLMVLEEGVIEGRRT 691...898
IV Seq. 9 AKLI 791...853 IACRCTPQQKADVALVIRK+ TGKRVCCIGDGGNDVSMIQCADVGVGI 899...902 KEGK+ASLAADFSITQFCHLTELLLWHGRNSY 933..1151
IV Seq. 14 CKLR 686...756 ICCRVSPLQKSEVVEMVKK+ VKVITLAIGDGANDVSMIQTAHVGVGI 802...805 NEGL+AANSSDYSIAQFKYLKNLLMVHGAWNY 836..1149
IV Seq. 11 SGLF 854...908 LCYRMTPSEKATIVNTVKK+ IKGNVLAIGDGANDVPMIQAAHVGIGI 954...957 KEGL+AAMACDFAIARFKFLSRLLLVHGHWSY 988..1454
V Seq. 4 CGII 1060..1730 VYARMKPKDKSDLILSLKK++ NNSYVGMCGDGANDCLALSCADIGISL 1777..1779 NNNE SSICSSFTSNKLCLHSIVHILIEGRAS 1809..1956
V Seq. 2 CGII 736...852 VFARMAPDQKQSLVEQLQQ IDYTVAMCGDGANDCAALKAAHAGISL 897...898 SDAE ASIAAPFTSKVPDIRCVPTVISEGRAA 928..1187
V Seq. 1 AGLI 1070..1159 IYARMSPDEKHELMIQLQK LDYTVGFCGDGANDCGALKAADVGISL 1204..1205 SEAE ASVAAPFTSKIFNISCVLDVIREGRAA 1235..1472
V Seq. 6 VGIV 714...788 VYARVSPSQKEFLLNTLKD MGYQTLMCGDGTNDVGALKQAHVGIAL 833...954 KLGD ASCAAPFTSKLANVSAVTNIIRQGRCA 984..1215
If Fig. 3 is not formatted perfectly by your browser try to click here.
Fig. 4.
Different phylogenetic trees produced from the dataset.
A total of 14 trees were constructed, in each case with a different input order of sequences into the Protdist and Fitch programmes. The number above the trees indicates how many times each tree occurred. Only major families are shown, indicated by hatched areas.
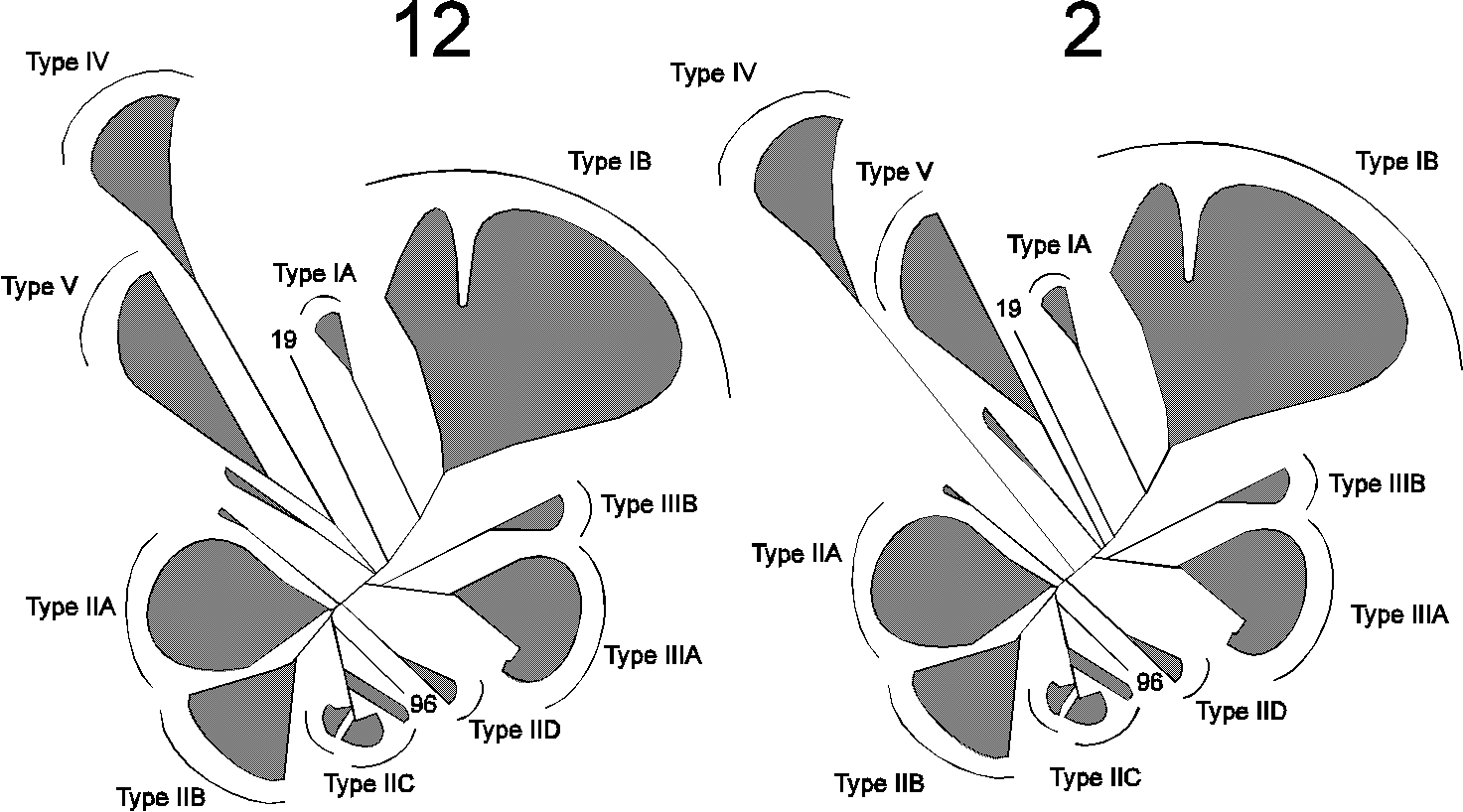
Fig. 5.
Consensus sequences in core segments of Type I - V P-type ATPases.
Repetitive lowercase letters on top indicate name of conserved segment. Amino acids identical in all members of given Type of P-type ATPases (Table 1) are in bold. The consensus amino acids shown in italic lowercase letters are positions where the sequences have one of two conserved amino acids I or V; D or E; F or Y. Numbers at top indicate the position of amino acids when conserved segments are arranged in a linear core sequence. Consensus sequences at top indicating amino acids present in 158, 135, and 90 sequences, respectively, are shown.
1 25 45 71 95
Segments aaaaaaaaaaaaaaaaaaaaaaaaa bbbbbbbbbbbbbbbbbbbb cccccccccccccccccccccccccc dddddddddddddddddddddddd
All 158/159 ---------------------D--- -------GE----------- -------------------------- ------------P-----------
All 135/159 -------GD------G--iP-D--- ------TGE----------- --------G---------G------- ------------P-----------
All 090/159 -----V-GDIv-v--Gd-iPAD--- vD-S-LTGES-PV-K----- f-G-----G-----V---G--T--G- ----v-i-V--iP--L---v----
IA 3/ 3 ----L---D---V--G--IP-DGE- VDESAITGESAPV--E-G-D TGGT---SD----------G--F-DR ---L-ALLV-LIPTTIG-LLSAIG
IB 23/35 -------GD---V-PG--i--DG-v -D-S--TGE--PV-K--G-- --G--N--G---v--------T---- ------VLvIACPCALGLATP---
IB 28/35 ------------v--G-----DG-v ------TGE--Pv------- --G-----G----------------- ------VLvi-CPC-L----P---
IB 35/35 ---------------------DG-- -------GE----------- -------------------------- ------------P-----------
IIA 17/26 -A-eLVPGDIV---VGD-VPAD-R- vd-S-LTGES--V-K--e-- F-GT----G----vVv-TG--TE-G- F-IAVALAVAAIPEGLPAViT-CL
IIA 21/26 -----VPGD-V----G--vPAD-R- v--S-LTGE---V-K----- f-GT----G----vV--TG--T--G- --i-V--AVA-IPEGLP-ViT--L
IIA 26/26 -------GD-v----G----AD--- -----L-GE---v-K----- f-------G---------------G- -------AV--iPEGLP--iT--L
IIB 8/12 -v-d--VGDI-----GD--PADGv- IDESSLTGESd-v-K--d-- LSGT-V-EGSG-M-VTAVGvNS--G- FII-VTv-VVAVPEGLPLAVT-SL
IIB 10/12 -v----VGDi-----GD--PADG-- iDESS-TGES--v-K----- -SGT-V-eG-G-M-vT-VG--S--G- FI--vTv-VVAVPEGLPLAVT--L
IIB 12/12 -------GDi-----G----AD--- ----S--GE----------- -SG-----G-----v--VG-----G- ----v-v--v-vPE-LPL-v---L
IIC 14/21 -AEieVVGD-VEvKGGDRiPAD-Ri VDNSSLTGESEPQ-RS-efT FFST---EGTA-GiVI-TGD-Tv-GR vIFLIGIIVANVPEGLLATVTVCL
IIC 17/21 -----V-GD-VEvK-GD-iPAD-Ri VDNSSLTGESEPQ-R--e-T FfST---EGT--GiVI--GD-Tv-GR viF-IGIIVANVPEGLLATVTV-L
IIC 21/21 -------G--V----G--iP---R- VD-SSLTGESe-Q------- -------e----G-vi--G-----G- -i-----iVA-VPEG---TvT---
IID 3/ 3 -S---V-GD------GD--PADLRL TDE-LLTGESLPV-KD---- -SSS-V-KGRA-GI---T-----IG- -IYA-----S-IP-SL--VL-ITM
IIIA 21/32 eA--LVPGDIi-i--G-IiPAD-RL iDQSALTGESLPV-K--GD- fSGST-K-GE---VV-ATG--TFFG- ----LV-LI-GIPIA-P-V---TM
IIIA 26/32 -A---VPGDI-----G-iiPAD-R- iDQSA-TGESL-V-K--Gd- fS-S--K-GE---vV-ATG--TF-G- ----L---I-GiPi--P-V---TM
IIIA 32/32 -----V-Gd------G-----D--- iD----TGES---------- ---S----GE----v--T---Tf-G- --------i---P-----V---T-
IIIB 3/ 3 PI--LVPGD---LAAGD--PAD-R- --Q--L-GESLPVEK----- -MGT-V-SG-AQA-V-ATG--TWFG- -LFAL-VAVGLTPEMLPMIV-S-L
IV 7/11 -W--v-VGDiV-i---d-IPAD-iL IET--LDGETNLK-R----- -d--LLRG--L-NT--v-GiVv-TG- ---fvILf---vPISL-V--E-iK
IV 9/11 ----v-VGD-v-------iPAD-iL i-T--LDGET--K------- ----------L-------G-Vv--G- -----I-f---vPISL-v--e--K
IV 11/11 -------GD-v---------AD--- i-T--LDGET--K------- --------------------V---G- -----------vP--L-v-----K
V 5/ 7 ---ELVPGDI---------PCD-iL V-E-MLTGESVPi-K----- --------------ViRTGF-T-KG- I---LDIIT--vPP-LP----v-i
V 6/ 7 ------P-DI---------PCD--- V-E--LTGESVP--K----- --------------V--TGF-T--G- ------I-T--vPP-LP-------
V 7/ 7 --------D------------D--- --E----GE-----K----- --------------V--T-f----G- ------I-----PP--P-------
96 131 188
Segments eeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeee fffffffffffffffffffffffffffffffffffffffffffffffffffffffff
All 158/159 ---------------------DKTGT---------- ----------------------------------------GD---------------
All 135/159 ----------E----------DKTGTLT-------- ---------------D---------i------G------TGD----A-------G--
All 090/159 ---V----AvE-LG----iCSDKTGTLT-N---V-- ---------G-----DPPR------i------GI-V-MiTGD---TA-AIA---Gi-
IA 3/ 3 NV-ATSGRAVEA-GD---L-LDKTGTITLGNR-A-- ---------GV--LKDI-K-GI-E-F---R-M---TVM-TGDN--TA--IA-EAGVD
IB 23/35 GiLiK----LE-------v-FDKTGTLT-G---V-- ------------Av-D---------i--L---G-----LTGDN---A-AiA---GI-
IB 28/35 GiL-K----LE-------v--DKTGTLT-G---V-- ---------------D---------i--L---G------TGD----A--iA---G--
IB 35/35 ---------------------DKTGT---------- ----------------------------------------GD---------------
IIA 17/26 NAiVR-LPSVETLGC--VICSDKTGTLTTN-M-V-- --E--L---G-vG--DPPR-EV--AI--C--AGIRV-MITGD---TA-AI---IG--
IIA 21/26 -A--R-LP-VETLG---VICSDKTGTLT-N-M-V-- -----L---G--G--DPPR--V---i--C--AGI-v--ITGD---TA-AI----G--
IIA 26/26 ------L--VE-LG----iC-DKTGTLT---M---- ---------G-----D--R--------------i-----TGD---TA--i-------
IIB 8/12 NNLVRHL-ACETMG-AT-ICSDKTGTLT-N-MTVV- -----LT-I--VGI-DPVRPEVP-A---C--AGITVRMVTGDNI-TARAIA--CGIL
IIB 10/12 -NLVR-L--CETMG-AT-ICSDKTGTLT-N-M-VV- ------------GI-DP-R--V--A---C--AGI-VRMVTGDN--TA-AIA--C-I-
IIB 12/12 --LVR----CETM------C-DKTGTLT-N-M---- ------------GI-D--R--V------C--AG--VRMV-GDN--TA--IA--C-I-
IIC 14/21 NCLVKNLEAVETLGSTS-ICSDKTGTLTQNRMTVAH FP---LCFVGL-SMIDPPRA-VPDAV-KCRSAGIKVIMVTGDHPITAKAIAK-VGII
IIC 17/21 NCLVKNLEAVETLGSTS-ICSDKTGTLTQNRMTVAH FP---L-F-GL-SMIDPPR--VPDAV-KCRSAGIKVIMVTGDHPITAKAIA--VGII
IIC 21/21 ------L--vETLGS---I-SDKTGTLTQNRMTV-H F------F--L-S---PPR--V--AV--C---GI-ViMVTGDHPITA-AIA--V-II
IID 3/ 3 -V-VR-L--LEALG-V-DICSDKTGT-TQG-M--R- --E--L-F--L-GIYDPPR-E--GAV---H-AGI-VHMLTGD---TAKAIA-EVGI-
IIIA 21/32 -AI-----AIEE-AG-dvLCSDKTGTLTLNKLSv-- ---G-W---G--P--DPPRHD-AeTI--A--LGv-VKMiTGD---I-KET-R-LGMG
IIIA 26/32 -AI-----AIE--AG-dvLCSDKTGTLT-NKL---- -----W---G--P--DPPR-D---TI--A--LG--VKM-TGD---I-KET-R-LG-G
IIIA 32/32 --I------iE-------LC-DKTGTLT-N------ -----W---------DPPR-D----i------G--vKM-TGD---I-------L---
IIIB 3/ 3 KVIVK-L-AIQNFGAMD-LCTDKTGTLTQD-I-LE- -DE--L--EG---FLDPPKE----A--AL---G--VK-LTGD---V-A--C-EVG-D
IV 7/11 ----R---I-EELGQ-EYIFSDKTGTLT-N-M-FKK -IE--L-LLG-T-IED-LQDGV-d-IE-L--AGIKiW-LTGDK-ETAINIG-S--L-
IV 9/11 ----------eeLG---Yi-SDKTGTLT-N-M-F-K --E--L-L-G-T-iED-LQ--V------L--AGI--W-LTGDK-ETA--I--S--L-
IV 11/11 -----------eLG-------DKTGTLT-N-M---- --E----L---T-iED-LQ--V---------A-I--W-LTGD--ETA--i-------
V 5/ 7 -IfC--P--i---G-I---CFDKTGTLTED-L---G --E--L-FLGF-i--N-LK--T---I--L-------iMiTGDN-LT---V--E-GIi
V 6/ 7 -I-C--P------G-I---CFDKTGTLT-d-L---- --E--L---G--i-----K------I-----------M-TGDN-LT---V------i
V 7/ 7 -i----P------G-i----FDKTGTLT-------- --E------G--i-----K---------------------GDN--T---v-------
234 265
Segments gggggggggggggggggggggggggggggggggggggggggggggg hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh
All 158/159 ------P--K-----------------GDG-ND------------- -------------------------------
All 135/159 ------P--K---v-------------GDG-ND-P-L--A-iG--- ---------d---------------------
All 090/159 vFA---P--K--iV--LQ-----VAMTGDGVNDAPALK-ADiGIA- GTDvA--AAD-vL-d-----Iv-Av---R-i
IA 3/ 3 F-AEATPE-K---I---Q--G-LVAMTGDGTNDAPALAQA-V--AM GTQAA-EA-NMVDLDS-PTKLI--V-IGKQ-
IB 23/35 v-A---P-dK---i--L---G--VAMVGDGINDAPALA-A--GiA- GTDvA---ADi-L----L--v-----L---T
IB 28/35 ------P--K---i---------V-MVGDGiNDAP-LA-A--G-A- G-DvA---AD--L----L-------------
IB 35/35 ------P--K-----------------GDG-ND------------- -------------------------------
IIA 17/26 -F-R--P-HK--IV--L----ei-AMTGDGVNDAPALK-AdIGIAM GT-VAK-ASdMVL-DDNF-TIV-AV-EGR-I
IIA 21/26 -F-R--P-HK---V--L-------AMTGDGVNDAPALK-AdIGiAM GT-VAK-A-dMvL-DDNF-TIV-Av-EGR-I
IIA 26/26 ---R-----K---v----------AMTGDG-N---AL--------- ---V--------L-DD-F--i--A---G--i
IIB 8/12 VLARSSP-DK-TLV----d---VVAVTGDGTND-PALK-ADVGFAM GTdVAKEASDIIL-DDNF-SIV-AV-WGRNV
IIB 10/12 V-ARS-P-DK--LV--------VVAVTGDGTND-PALK-ADVG-AM GTdVAKeASDiI--DDNF--IV--v-WGR-V
IIB 12/12 V-AR--P-DK------------vVAVTGDG-ND-PAL-----G--M GT-VA-----ii---D-F--IV------R-v
IIC 14/21 VFARTSPQQKLIIVEGCQR-GAIVAVTGDGVNDSPALKKADIGVAM GSDVSKQAADMILLDDNFASIVTGVEEGRLI
IIC 17/21 VFARTSPQQKLIIVEGCQR---iVAVTGDGVNDSPALKKADIGvAM GSD--K-AADMILLDDNFASIVTGVEEGRLI
IIC 21/21 VFARTSP-QK--IVE--Q-----V-VTGDG-ND-PAL-KADIGvAM G-D--K--AD-iLL-DNFAS-V-GvE-GR-I
IID 3/ 3 VI-RC-PQTKV-MIEALHRR--F--MTGDGVNDSPSLK-ANVGI-M GSDV-K-ASDIVL-DDNF-SI-NA-EEGRRM
IIIA 21/32 GFA-VFPEHKY-IV--LQ-R---V-MTGDGVNDAPALKKAD-GIAV ATDAAR-A-DIVLT-PGLS-II-A--TSR-I
IIIA 26/32 GFA-VFP-HKY-iV--LQ-------MTGDGVNDAP-LKKAD-GIAV ATDAAR-A-DIV---PGLS-II-A---SR-I
IIIA 32/32 GF----P--Ky--------------MTGDGVND-P-LK-A--GiA- --DAAR---D-V----G---ii-A----R-i
IIIB 3/ 3 -FARLTP--K-RI---L---GH-VGF-GDGINDAPALR-AD-GISV A-DIA-E--DIILLEK-LMVLEEGVI-GR-T
IV 7/11 iCCR-SP-QKA-vV--v-------LAIGDG-NDV-MIQ-A-VGvGI -EG-A----DY-I-QF--L--LLLVHGR--Y
IV 9/11 -CCR--P-QKA-vV--v--------AIGDG-NDV-MIQ-A-vGvGI -EG-A----Dy-I-QF-----L-LVHG---Y
IV 11/11 ---R--P--K----------------I--G-ND--MI--A--GvGI -EG-A----D-----F-----L---HG---Y
V 5/ 7 VyARM-P-QK--Li--L------V-MCGDGANDC-ALK-A-vGISL ---eASiAAPFTS----i--V--VI-EGR--
V 6/ 7 VyAR--P--K------L--------MCGDG-ND--AL--A--GI-L ---e-S----FTS-------v--v---GR--
V 7/ 7 vyAR--P--K------L----------GDG-ND--A---A--Gi-- ---e------f-S-------v--v---GR--
If Fig. 5 is not formatted perfectly by your browser try to click here.
Fig. 6.
Phylogenetic tree based on core sequences of 159 P-type ATPases.
The tree was constructed using the neighbour joining method. Some areas (which are not connected to the rest of the tree) have been enlarged 40 % to clarify the distribution of species. When the substrate specificity of the ATPases present in each family is known, it corresponds in all cases to the name of the family. The numbers of the sequences correspond to the numbers used in Table 1. The black branches show ATPases originating from bacteria and archaea, and the grey branches show ATPases originating from eukarya. The P-type ATPases from the fully sequenced organisms are shown with the following symbols:  : Escherichia coli;
: Escherichia coli;  : Haemophilus influenzae;
: Haemophilus influenzae;  : Methanococcus jannaschii;
: Methanococcus jannaschii;  : Mycoplasma genitalium;
: Mycoplasma genitalium;  : Mycoplasma pneumoniae;
: Mycoplasma pneumoniae;  : Saccharomyces cerevisiae;
: Saccharomyces cerevisiae;  : Synechocystis PCC6803. The abbreviations are HM: heavy metals; NAS: no assigned specificity; PL: phospholipids.
: Synechocystis PCC6803. The abbreviations are HM: heavy metals; NAS: no assigned specificity; PL: phospholipids.