| [1] |
A. Aitken, Trends Cell Biol. 6 (1996) 341-347. |
| [2] |
K. Altendorf, A. Siebers, W. Epstein, Ann. N.Y. Acad. Sci. 671 (1992) 228-243. |
| [3] |
I.T. Arkin, P.D. Adams, A.T. Brunger, S.O. Smith, D.M. Engelman, Ann. Rev. Biophys. Biomol. Struct. 26 (1997) 157-179. |
| [4] |
K.B. Axelsen, M.G. Palmgren, J. Mol. Evol. 46 (1998) 84-101. |
| [5] |
L. Baunsgaard, A.T. Fuglsang, T. Jahn, H.A.A.J. Korthout, A.H. de Boer, M.G. Palmgren, Plant J. 13 (1998) 661-667. |
| [6] |
P. Beguin, M.C. Peitsch, K. Geering, Biochemistry 35 (1996) 14098-14108. |
| [7] |
P. Beguin, X.Y. Wang, D. Firsov, A. Puoti, D. Claeys, J.D. Horisberger, K. Geering, EMBO J. 16 (1997) 4250-4260. |
| [8] |
F.R. Blattner et al., Science 277 (1997) 1453-1474. |
| [9] |
C.J. Bult et al., Science 273 (1996) 1058-1073. |
| [10] |
E.T. Buurman, K.T. Kim, W. Epstein, J. Biol. Chem. 270 (1995) 6678-6685. |
| [11] |
P. Catty, A.D. D'Exaerde, A. Goffeau, FEBS Lett. 409 (1997) 325-332. |
| [12] |
N. Courtois-Coutry, D. Roush, V. Rajendran, J.B. McCarthy, J. Geibel, M. Kashgarian, M.J. Caplan, Cell 90 (1997) 501-510. |
| [13] |
K.W. Cunningham, G.R. Fink, J. Cell Biol. 124 (1994) 351-363. |
| [14] |
L.A. Dunbar, D.L. Roush, N. Courtois-Coutry, T.R. Muth, C.J. Gottardi, V. Rajendran, J. Geibel, M. Kashgarian, M.J. Caplan, Ann. N.Y. Acad. Sci. 834 (1997) 514-523. |
| [15] |
A. Enyedi, T. Vorherr, P. James, D.J. McCormick, A.G. Filoteo, E. Carafoli, J.T. Penniston, J. Biol. Chem. 264 (1989) 12313-12321. |
| [16] |
P. Eraso, F. Portillo, J. Biol. Chem. 269 (1994) 10393-10399. |
| [17] |
R. Falchetto, T. Vorherr, J. Brunner, E. Carafoli, J. Biol. Chem. 266 (1991) 2930-2936. |
| [18] |
M.J. Fagan, M.H. Saier Jr., J. Mol. Evol. 38 (1994) 57-99. |
| [19] |
R. Falchetto, T. Vorherr, E. Carafoli, Protein Sci. 1 (1992) 1613-1621. |
| [20] |
N. Ferrol, A.B. Bennett, Plant Cell 8 (1996) 1159-1169. |
| [21] |
M.S. Feschenko, K.J. Sweadner, J. Biol. Chem. 272 (1997) 17726-17733. |
| [22] |
M.S. Feschenko, R.K. Wetzel, K.J. Sweadner, Ann. N.Y. Acad. Sci. 834 (1997) 479-488. |
| [23] |
G. Fisone et al., J. Biol. Chem. 269 (1994) 9368-9373. |
| [24] |
R.D. Fleischmann et al., Science 269 (1995) 496-512. |
| [25] |
C.M. Fraser et al., Science 270 (1995) 397-403. |
| [26] |
C.M. Fraser et al., Nature 390 (1995) 580-586. |
| [27] |
K. Geering, A. Beggah, P. Good, S. Girardet, S. Roy, D. Schaer, P. Jaunin, J. Cell Biol. 133 (1996) 1193-1204. |
| [28] |
A. Goffeau et al., Nature 387(Suppl.) (1997) 5-105. |
| [29] |
R.M. Gopinath, F.F. Vincenci, Biochem. Biophys. Res. Commun. 77 (1977) 1203-1209. |
| [30] |
J.F. Harper, B. Hong, I. Hwang, H.Q. Guo, R. Stoddard, J.F. Huang, M.G. Palmgren, H. Sze, J. Biol. Chem. 273 (1998) 1099-1106. |
| [31] |
Hebsgaard, S.M., Korning, P.G., Tolstrup, N., Engelbrecht, J., Rouze, P. and Brunak, S. (1996) Nucleic Acids Res. 24, 3439-3452. |
| [32] |
R. Himmelreich, H. Hilbert, H. Plagens, E. Pirkl, B.C. Li, R. Herrmann, Nucleic Acids Res. 24 (1996) 4420-4449. |
| [33] |
L. Huang, T. Berkelman, A.E. Franklin, N.E. Hoffman, Proc. Natl. Acad. Sci. USA 90 (1993) 10066-10070. |
| [34] |
T. Jahn, A.T. Fuglsang, A. Olsson, I.M. Brüntrup, D.B. Collinge, D. Volkmann, M. Sommarin, M.G. Palmgren, C. Larsson, Plant Cell 9 (1997) 1805-1814. |
| [35] |
P. James, M. Maeda, R. Fischer, A.K. Verma, J. Krebs, J.T. Penniston, E. Carafoli, J. Biol. Chem. 263 (1988) 2905-2910. |
| [36] |
H.W. Jarrett, J.T. Penniston, Biochem. Biophys. Res. Commun. 77 (1977) 1210-1216. |
| [37] |
P.L. Jørgensen, J.P. Andersen, J. Membr. Biol. 103 (1988) 95-120. |
| [38] |
P.L. Jørgensen, J.H. Rasmussen, J.M. Nielsen, P.A. Pedersen, Ann. N.Y. Acad. Sci. 834 (1997) 161-174. |
| [39] |
T. Kaneko et al., DNA Res. 3 (1996) 109-136. |
| [40] |
H.-P. Klenk et al., Nature 390 (1997) 364-370. |
| [41] |
F. Kunst et al., Nature 390 (1997) 249-256. |
| [42] |
J.B. Lingrel, J.M. Argüello, J. van Huysse, T.A. Kuntzweiler, Ann. N.Y. Acad. Sci. 834 (1997) 194-206. |
| [43] |
N.S. Logvinenko, I. Dulubova, N. Fedosova, S.H. Larsson, A.C. Nairn, M. Esmann, P. Greengard, A. Aperia, Proc. Natl. Acad. Sci. U.S.A. 93 (1996) 9132-9137. |
| [44] |
S. Lutsenko, J.H. Kaplan, Biochemistry 34 (1995) 15607-15612. |
| [45] |
S. Lutsenko, K. Petrukhin, T.C. Gilliam, J.H. Kaplan, Ann. N.Y. Acad. Sci. 834 (1997) 155-157. |
| [46] |
D.H. MacLennan, W.J. Rice, A. Odermatt, Ann. N.Y. Acad. Sci. 834 (1997) 175-185. |
| [47] |
S. Malmström, P. Askerlund, M.G. Palmgren, FEBS Lett. 400 (1997) 324-328. |
| [48] |
J. Moniakis, M.B. Coukell, A. Forer, J. Biol. Chem. 270 (1995) 28276-28281. |
| [49] |
P. Morsomme, A. de Kerchove d'Exaerde, S. De Meester, D. Thinès, A. Goffeau, M. Boutry, EMBO J. 15 (1996) 5513-526. |
| [50] |
A.J. Muslin, J.W. Tanner, P.M. Allen, A.S. Shaw, Cell 84 (1996) 889-897. |
| [51] |
J.V. Møller, B. Juul, M. le Maire, Biochim. Biophys. Acta 1286 (1996) 1-51. |
| [52] |
C. Navarre, P. Catty, S. Leterme, F. Dietrich, A. Goffeau, J. Biol. Chem. 269 (1994) 21262-21268. |
| [53] |
C. Oecking, M. Piotrowski, J. Hagemeier, K. Hagemann, Plant J. 12 (1997) 441-453. |
| [54] |
M.G. Palmgren, Adv. Bot. Res. 28 (1998) 1-70. |
| [55] |
M.G. Palmgren, M. Sommarin, R. Serrano, C. Larsson, J. Biol. Chem. 266 (1991) 20470-20475. |
| [56] |
F. Portillo, I.F. de Larrinoa, R Serrano, FEBS Lett. 247 (1989) 381-385. |
| [57] |
F. Portillo, P. Eraso, R. Serrano, FEBS Lett. 287 (1991) 71-74. |
| [58] |
B. Regenberg, J.M. Villalba, F.C. Lanfermeier, M.G. Palmgren, Plant Cell 7 (1995) 1655-1666. |
| [59] |
R. Serrano, FEBS Lett. 156 (1983) 11-14. |
| [60] |
D.R. Smith et al., J. Bacteriol. 179 (1997) 7135-7155. |
| [61] |
A. Sorin, G. Rosas, R. Rao, J. Biol. Chem. 272 (1997) 9895-9901. |
| [62] |
K.T. Togawa, T. Ishiguro, K. Kaya, A. Shimada, T. Imagawa, K. Taniguchi, J. Biol. Chem. 270 (1995) 15475-15478. |
| [63] |
J.F. Tomb et al., Nature 388 (1997) 539-547. |
| [64] |
A.K. Verma, A. Enyedi, A.G. Filoteo, J.T. Penniston, J. Biol. Chem. 269 (1994) 1687-1691. |
| [65] |
B. Vilsen, D. Ramlov, J.P. Andersen, Ann. N.Y. Acad. Sci. 834 (1997) 297-309. |
| [66] |
T. Vorherr, M. Chiesi, R. Schwaller, E. Carafoli, Biochemistry 31 (1992) 371-376. |
| [67] |
M.B. Yaffe, K. Rittinger, S. Volinia, P.R. Caron, A. Aitken, H. Leffers, S.J. Gamblin, S.J. Smerdon, L.C. Cantley, Cell 91 (1997) 961-971. |
| [68]
| M. Zurini, J. Krebs, J.T. Penniston, E. Carafoli, J. Biol. Chem. 259 (1984) 618-627. |
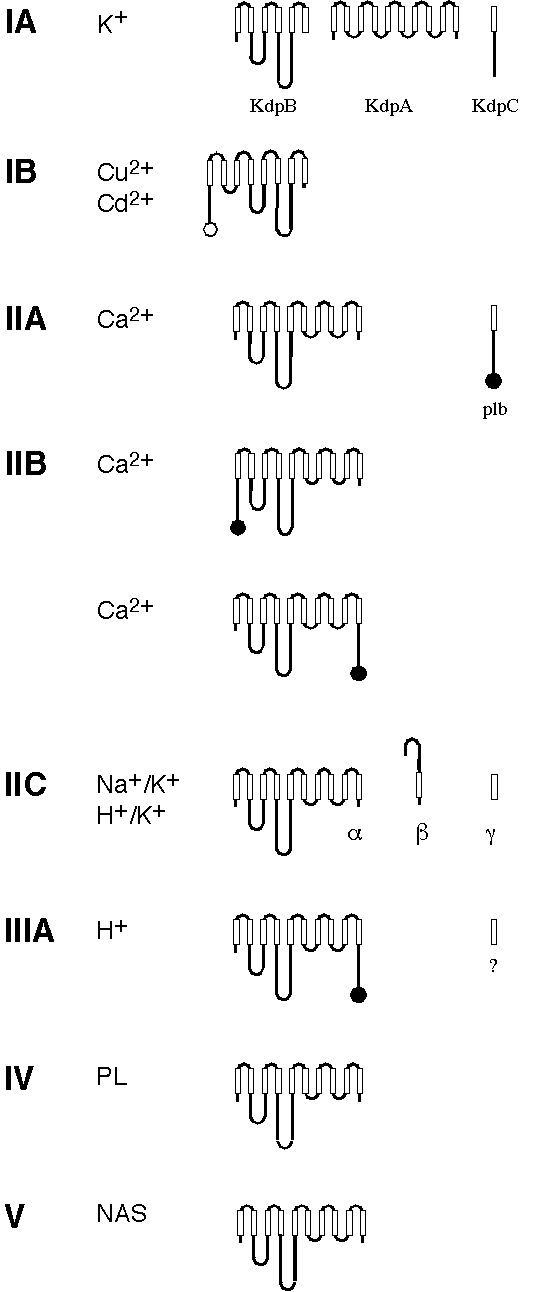
 subunit in addition to the catalytic
subunit in addition to the catalytic  subunit. The
subunit. The 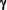 subunit, which seems to be important for modulating K+ activation of the pump [7]. The plasma membrane H+-ATPase (type IIIA) of S. cerevisiae is likewise associated with a small proteolipid for which two genes exist, PMP1 and PMP2 [52]. The function of this component remains obscure.
subunit, which seems to be important for modulating K+ activation of the pump [7]. The plasma membrane H+-ATPase (type IIIA) of S. cerevisiae is likewise associated with a small proteolipid for which two genes exist, PMP1 and PMP2 [52]. The function of this component remains obscure.